HUMAN DIVERSITY
Modes of evolution Throughout course we have stressed that traits must have a genetic basis
WHY? for transmission to next generation. What do humans have
that allows transmission to next generation without genetics?? CULTURAL
transmission. This is a major distinction with other animals (although
other animals do have "culture", it is not as "advanced"
as humans) Cultural transmission can be vertical (between generations)
or horizontal (within generation). Vertical cultural transmission is
Lamarkian: what you acquire during your lifetime you can pass on
to your offspring ("inheritance of acquired characteristics").
Some analogies with other evolutionary forces:
Communication, and especially language, are further "key innovations" that may have lead humans into a new "adaptive zone". But literature (Lascaux cave paintings, National Inquirer; and now consider electronic transfer of literary info, e-mail, etc.) really sets us apart. Our rate of evolution is dramatic on all counts. While the beaver, in building its dam, alters the environment that surrounds it, it has evolved in this context of altering its environment in a predictable way for a long time. We are no where near "equilibrium" with respect to how we are evolving with the extensive alterations we are making to our environment. One hopes that we have the genetic wherewithal to "run fast enough just to stay in place" (as the Red Queen suggests we must).
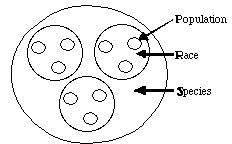
All organisms vary and humans are quite good at recognizing nodes or
clusters in that variation (demes, populations, species). The nodes or
clusters within human phenotypic variation are quite pronounced and most
of us would sort the ambassadors to the UN into more-or-less the same "groups".
The question thus arises: how is the genetic variation within humans partitioned?
This question was asked (and answered) by R. C. Lewontin in 1972 (The apportionment
of human diversity. Evolutionary Biology vol. 6: pp. 381-398).
Lewontin collected data on the frequencies of different alleles at various
nuclear loci in different human populations. These populations were
nested into larger groups we know as races, and the various races
can be nested further into the larger group we call the species Homo
sapiens.
With data on allele frequencies in each population, Lewontin could ask
how much additional variation is added to the unit in question by pooling
together the data from all populations within races, or at one level up
the hierarchy, by pooling all the data from within races to one large species
sample.
To quantify what proportion of the total genetic variation within humans,
a measure of "heterozygosity" was used similar to H = 2pq (or
[1- (allele frequency i)2]
for i different alleles; in the paper the very similar Shannon-Weaver index
was used). The numbers obtained from this formula provide a measure of
genetic diversity at each of the many loci tabulated by Lewontin. Four
points are relevant in thinking about this measure of "diversity":
1) a locus with only one allele will have H = 0; 2) the greatest diversity
will be when all alleles are equally frequent (p=q=0.5 for 2 alleles; p1=p2=p3=p4=0.25
for 4 alleles); 3) diversity will increase as the number of alleles increases
(the 4 allele case above is more 'diverse' than the 2 allele case); 4)
diversity is a convex function of allele frequency (a diversity
measure from a pooled sample obtained by combining alleles from two different
populations will be greater than the average of the two diversity measures
from each population).
These H values can be tabulated for several different levels of the
hierarchy described above: by considering a single population's p and q,
or the average p and average q within a single race (averaged among all
populations within that race = Hpop), or
the average p and average q for all races (pooling all populations within
a race to determine the p for that race, then averaging the p's for each
of the races = Hrace). Hspecies
will be determined by pooling all populations irrespective of race to obtain
a species-wide p, then calculating H = 2pq (or as above for the multiple
allele case). Thus we can have an Hpop
which will be less than Hrace which, in
turn, will be less than Hspecies.
These H values can then be partitioned or apportioned so that
the total variation within humans can be attributed to the within population
component or to the within race component or to the between
race component. The simple expressions for these, and the data for
each locus are presented below. The conclusions from the results are very
clear. Of all the variation within humans, 85.4% of it lies within
populations (i.e. is due to variation among individuals within populations).
An additional 8.3% lies between populations within races. Only
6.3% of all the genetic variation within humans is due to differences
between races!
Recent analyses with microsatellites in human populations give slightly
different numbers, but the general conclusions are the same. Microsatellites
are regions of the genome that generally show a repetition of a simple
sequence, such as CA repeated over and over. In some instances the repeated
units can be longer and these regions are called minisatellites. See figs.
10.9, 10.10, pg. 271-272 for an example. Such regions are very useful since
the repeated sequence allows for the insertion and deletion of repeats.
As a result, there can be many alleles in a population that differ
in the number of repeated units in the specific region of the chromosome.
This allows for the discrimination among individuals, based on whether
they share, or do not share, alleles of similar length. This is determined
by amplifying a person's DNA using specific primers in a PCR reaction,
and running the two samples out on a gel. If bands are shared, the two
individuals are related, if the band sizes do not match, the two are unrelated
(or if you are an accused criminal, you might be convicted or let off the
hook based on these sorts of DNA 'fingerprint' analyses). As with protein
allele frequencies, one can still calculate the H values described above
and partition the variation into different levels of a hierarchy.
Lewontin concludes that there is no genetic or taxonomic basis to racial distinction and classifications of this sort are of no social value. While you are free to agree or disagree with Lewontin's social interpretation of the data, the population genetic conclusions are clear: with the largest component due to variation among individuals within populations, each and every one of us matters.