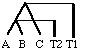MOLECULAR EVOLUTION
For evolutionists the revolution in DNA technology has been a major
advance. The reason is that the very nature of DNA allows it to be used
as a "document" of evolutionary history: comparisons of the DNA
sequences of various genes between different organisms can tell us a lot
about the relationships of organisms that cannot be correctly inferred
from morphology. One definite problem is that the DNA itself is a scattered
and fragmentary "document" of history and we have to beware of
the effects of changes in the genome that can bias our picture of organismal
evolution.
Two general approaches to molecular evolution are to 1) use DNA to study
the evolution of organisms (such as population structure, geographic variation
and systematics) and to 2) to use different organisms to study the evolution
of DNA. To the hard-core molecular evolutionist of the latter type, organisms
are just another source of DNA. Our general goal in all this is to infer
process from pattern and this applies to the processes of organismal
evolution deduced from patterns of DNA variation, and processes of molecular
evolution inferred from the patterns of variation in the DNA itself. An
important issue is that there are processes of DNA change within the
genome that can alter the picture we infer about both organismal and
DNA evolution: the genome is fluid and some of the very processes
that make genomes "fluid" are of great interest to evolutionary
biologists. Thus molecular evolution might be called the "natural
history of DNA". The points that follow are some interesting observations
interspersed with some basic concepts.
Some important background: DNA has many different roles in terms of
function. Most of our DNA does not code for proteins (more below) and thus
is quite a different type of character/trait than DNA that does code for
protein. In eukaryotes, genes are frequently broken up into exons
(expressed) and introns (spliced out of the RNA before becoming
a true messenger RNA). The genes also have regulatory sequences that indicate
when and where to transcribe the DNA into RNA for protein synthesis. The
genetic code is the information system for translating the sequence
of RNA into the sequence of amino acids. Within this triplet code
some of the nucleotide positions are silent or synonymous because
any nucleotide in that position will do (see table 2.1 pg. 25). This "universal"
code is not completely universal because the mitochondrial genome uses
some of the codons in different ways (e.g., some termination codons in
the universal code specify amino acids in the mitochondrial code). Thus
even the genetic code can evolve.
Some important theoretical background: we want to develop a picture of what happens to a new mutant in a population, lets say a single nucleotide change a one position in the DNA. This is the starting point for molecular evolution. If the new mutant is governed by genetic drift, its fate should be quite different than another nucleotide mutation that is governed by selection (see below). To describe molecular evolution Kimura formulated the Neutral theory of molecular evolution which is remarkably simple. If:
u = mutation rate / gene / generation, N = population size, then the
number of new mutations occurring per generation in a population = 2Nu
(2 because we are considering diploid organisms).
Now, when a new mutation occurs in a population its initial frequency
= 1/2N because it is the one new variant out of a total of 2N genes
in the population. This is also its probability of fixation because
the probability of you reaching into a barrel of 2N marbles and getting
the one new marble is 1/2N. Thus taking these two values (the number of
new mutations/generation and the probability of fixation), the rate
of substitution, K is just their product: 2Nu 1/2N = u (substitution
means that the new mutant goes to fixation in the population and substitutes
the original nucleotide or gene). Kimura got very famous for this simple
bit of algebra. It tells us that the neutral rate of molecular evolution
is equal to the neutral mutation rate. Now a question arises: what is the
distribution of the types of mutations? Are most neutral? Are some deleterious?;
beneficial? (see figure 7.1, pg. 153 and box 7.2, pg. 176-177). One consequence
of the neutral theory is that genes with different mutation rates will
have different rates of evolution.
The rate of evolution of a gene or mutation that is under selection
will be very different. Similarly different genes with different
functions, or different parts of a gene with different functions
will have different rates of evolution. Thus, different regions of DNA
with different functional constraints will evolve at different rates
(see table 7.1, pg. 156; table 7.7, pg. 183).
One prediction of the neutral theory is that silent (synonymous)
sites in protein coding regions will evolve faster than replacement (nonsynonymous)
sites (due to different functional constraints). This provides a null
hypothesis about DNA evolution. Most sequences fit this neutral model;
however, the histocompatibility loci appear to deviate from a neutral model
in that there are more nonsynonymous substitutions than synonymous
substitutions. This holds only for the antigen binding region; the rest
of the molecule is consistent with neutral expectations. (see figure 7.10,
pg. 185).
Another prediction of the neutral theory is that amount of sequence
divergence will be correlated with the level of heterozygosity; heterozygosity
is measured as 2pq for a two allele situation or (2pq+2pr+2qr) for a three
allele situation, or 1-xi2
for i alleles; see figure 5.2 pg. 96 for a two allele view). Loci with
high heterozygosity should evolve at a faster rate under the assumption
that these loci have a higher rate of neutral mutation (thus more variation
within species and more substitution between species; se Kimura's proof
above). In general, loci fit this relationship, however, balancing selection
at a locus will introduce more heterozygosity then expected. What about
purifying selection?
Different rates of substitution have also been observed in different lineages of organisms: For the human - chimp divergence, the rate = 1.3 x10-9 substitutions/nucleotide site/year
for the human - Old World monkey split, the rate = 2.2x10-9 substitution/site/year
for the mouse - rat split (=rodents), the rate = 7.9x10-9
substitutions/site/year. Thus, rodents appear to have a faster rate of
molecular evolution. It has been argued that a shorter generation time
in rodents accounts for the faster rate of evolution, the so called generation
time effect. (see table 7.5, pg. 179). There are examples where short
generation species have slower rates of evolution; the point is that rates
differ, the cause(s) of these rate differences have not been unambiguously
identified.
Most discussions of the rates of DNA evolution have been with respect
to the molecular clock hypothesis which states that there is a positive
linear relationship between time since two species diverged and
amount of genetic divergence (e.g., DNA sequence difference) between
those species. These observations stated above indicate that there is not
one molecular clock but probably many molecular clocks that
"tick" at different rates.
Lets say we identify a reliable molecular clock (e.g., number of amino
acid substitutions in the cytochrome C gene), we can use this to date,
or corroborate, evolutionary events of interest (e.g., the divergence times
for species that do not have good fossil data). For example: we know that
there are KXY substitutions between species
X and Y and we know that they diverged T years ago (from fossil data).
Thus the rate of molecular evolution is r = KXY/2T.
The denominator has a 2 in it because there are two paths of evolution
on which the divergence can accumulate (ancestor to X and ancestor to Y).
Now lets say we obtain the sequence of cytochrome C from species A, B and
C (see figure below). From these data we count up the number of substitutions
for all three pairwise comparisons (the K's). We are given the date of
divergence between A and C (=T1) and we want to date
the divergence of A and B (T2 = ?) but don't have
any fossil data. If one assumes that the rate of evolution is the same
in species A, B and C as it was measured to be between species X and Y,
we can use the amount of sequence divergence between species A,
B and C to estimates their dates of divergence (measured in millions
of years before present, MYBP). See box 7.1, pg. 172.
Repetitive DNA Studies of many organisms has revealed that a
large proportion of eukaryotic genomes consists of repetitive DNA.
Some of this is short localized repeats: in the kangaroo rat the sequence
(AAG) is repeated 2.4 billion times, the sequence (TTAGGG) is repeated
2.2 billion times and the sequence (ACACAGCGGG) is repeated 1.2
billion times. What it does is unclear. Sequences like this have
been called junk DNA. Note that junk is stuff you don't throw
away because it might be useful some day; garbage is stuff you don't want
so you throw it away. These sequences might have some function we don't
know about so they have been called junk DNA. The fact that such sequences
seem to accumulate in genomes has lead to the notion that repetitive DNA
is selfish DNA, since the sequence makes additional copies of itself
within the genome decoupled from the reproduction rate of the host (i.e.,
the kangaroo rat).
Another form of repetitive DNA are transposable elements. These
are sequences of DNA that generally code for certain proteins and have
the ability to move around the genome in a process called transposition.
There are quite a number of different types of such elements (we will not
review them all). The point is that they (like other repetitive DNA) are
governed by intragenomic dynamics as well as organismal population
dynamics. An example is the P element in Drosophila melanogaster.
There are strains of flies that have P elements (P strains) and strains
that do not (M strains). When a P male is crossed to an M female the P
elements enter the genome of the offspring and jump around causing mutations
(this is what we mean by a fluid genome). A curious observation
about P elements is that strains of flies collected from natural populations
before 1950 do not have P elements whereas flies collected from the wild
after that do have them. A variety of observations indicate that P elements
invaded D. melanogaster recently. The best evidence is that
D. melanogaster's close relative do not have P elements, but a more
distantly related fly D. willistoni does have them and they differ
by only a few nucleotides over 2900 base pairs of DNA. These data suggest
that P elements in D. melanogaster are the result of a horizontal
gene transfer (horizontal as opposed to "vertical" as one
inherits DNA from ones parents or ancestor "above"). Thus, not
only can DNA move around the fluid genome, but if DNA from one species
can enter the gene pool of another without the species fusing into one,
one has to be very aware of what DNA sequence one is using to determine
phylogenies, etc.
If multiple copies of a DNA sequence are present in a genome we can
think of each sequence as a single "species" evolving on its
own "line of descent" because each repeat will be mutated at
random. Thus if we had the complete DNA sequence of all the repeated P
elements within a genome, we would find that they are not identical and
thus we could build a cladogram of these elements much like we can build
a cladogram of birds. When this sort of analysis was done on different
kinds of repeated elements (many copies of ribosomal DNA, for example)
it was found that the copies showed almost no variation. This observation
suggested that all the repeats of this family (ribosomal DNA family) were
evolving in concert, i.e., together. This pattern of homogeneity
of repeats is called concerted evolution (figure 10.5, pg. 262).
The process(es) that generate this pattern could be unequal crossing
over (see figure 10.2 & 10.6, pg. 258, 263) or gene conversion.
Gene conversion is when the sequence of one region of DNA is used as
a "template" to "correct" or modify the sequence of
another region of DNA. We do not need to go into the molecular details
of these processes, but that DNA can evolve in concert with other sequences
in the genome again indicates that intragenomic dynamics can influence
the pattern of DNA variation we see within and between species.
Gene duplication is a minimalist version of repetitive DNA (figures
10.1-10.3, pgs. 257-259). Many genes in the genome are duplicated and when
this happens one of the copies may be "freed" from constraints
and evolve a new function. The best understood case of this phenomenon
is the evolution of the globin genes myoglobin, a-hemoglobin,
ß-hemoglobin. The existence of duplicated genes forces us to recognize
different kinds of homology because there are two ways to have a
common ancestor: by gene duplication and by speciation. When
two genes share a common ancestor due to a duplication event we call them
paralogous (a-hemoglobin
and ß-hemoglobin in you are paralogous as are the a-hemoglobin
in you and the ß-hemoglobin in chimps). When two genes share a common
ancestor due to a speciation event we call them orthologous (a-hemoglobin
in you and a-hemoglobin
in chimps). Obviously when constructing a cladogram from molecular data
one should use orthologous genes if one wants to build a tree of organisms.
A further example of the fluid genome is exon shuffling This
is the pattern observed when exons or functional domains
of genes are shuffled together to form new or modified genes. Some genes
have very distinct domains that have clear relationships to other domains
in very different genes. It is thought that these domains have been moved
around the genome by transposition or illegitimate recombination events
in evolution, accidentally forming new associations that happen
to have novel functions. Wally Gilbert proposed the idea of exon shuffling
and argued that such a phenomenon might accelerate evolution by creating
new material for adaptive evolution.
One of the more interesting observations to ponder in molecular evolution
is the C-value paradox. The C value of a species is the Characteristic
or Constant amount of DNA in a haploid genome of that species.
If we look at the diversity of organisms from viruses to humans we see
a clear trend in biological complexity. If we compare the C values across
this range of organisms (assuming viruses are "organisms") some
of the less complex organisms have much more DNA than the more complex
organisms (see tables 10.2, 10.3, pgs. 259-260). This presents a paradox.
If DNA codes for proteins that give us form and function, what is a lowly
alga doing with all that DNA? We just don't know. Presumable most of it
is not "functional" in the sense of coding for proteins and RNAs.
Much of it may be "junk DNA", but maybe this "junk"
helps in aligning chromosomes properly during mitosis an meiosis.
You now see why we referred to this topic as the natural history of DNA: these are "stories" we know about how DNA "behaves" in evolution. The general point is that the intragenomic dynamics of DNA and the intergenomic (Åinterorganism) dynamics can be radically different, but the patterns of variation we see in the current day are the summed effects of both processes. This means we have to know some things about a piece of DNA before we can use it as an evolutionary tool.